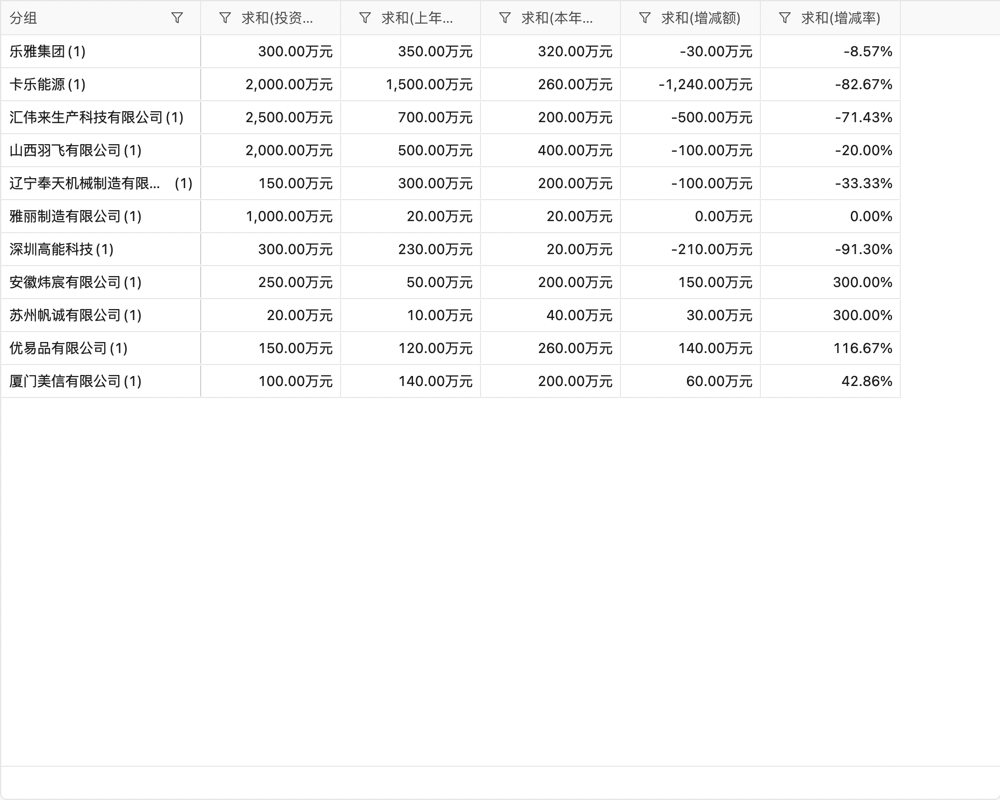
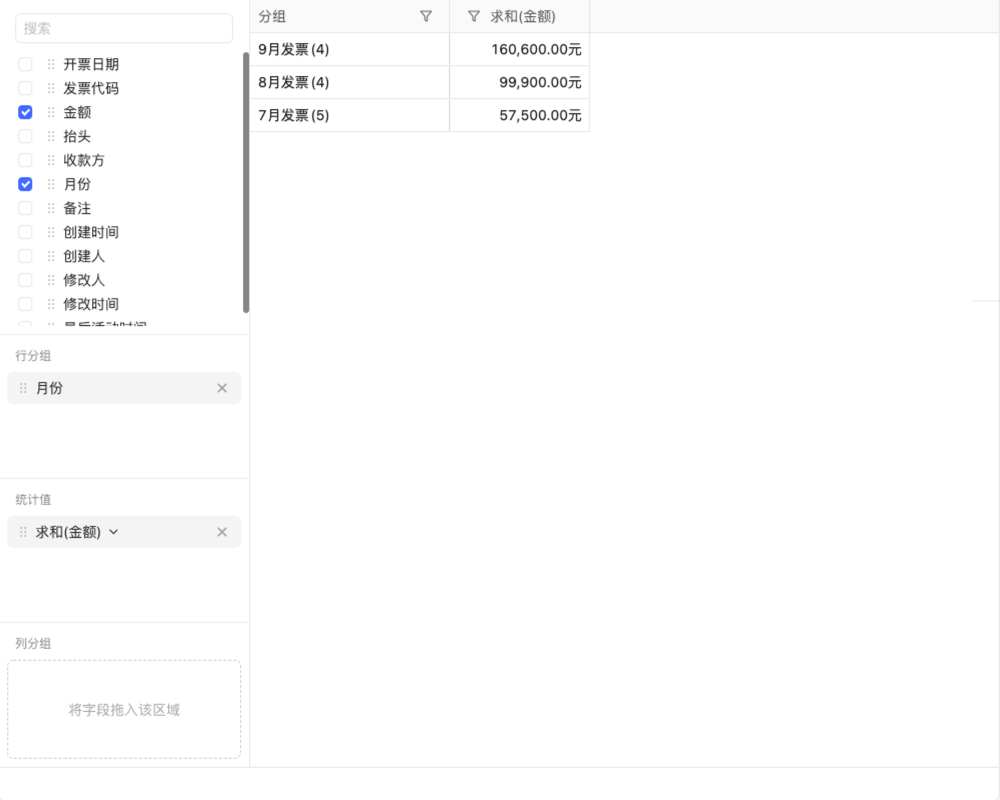
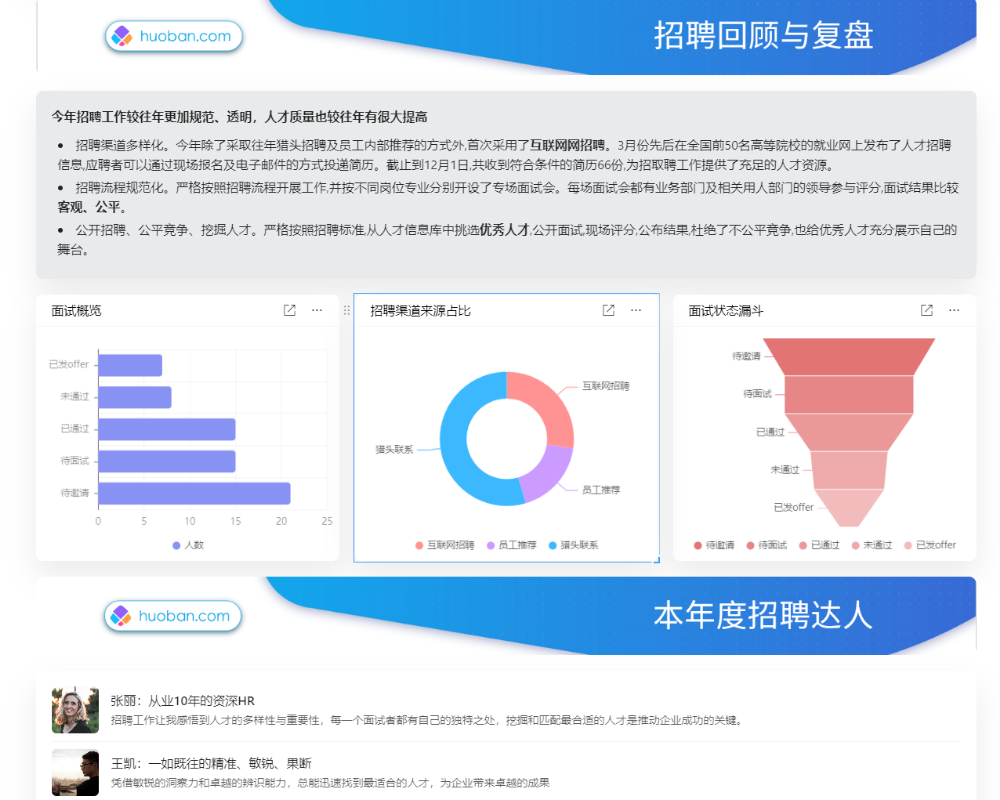
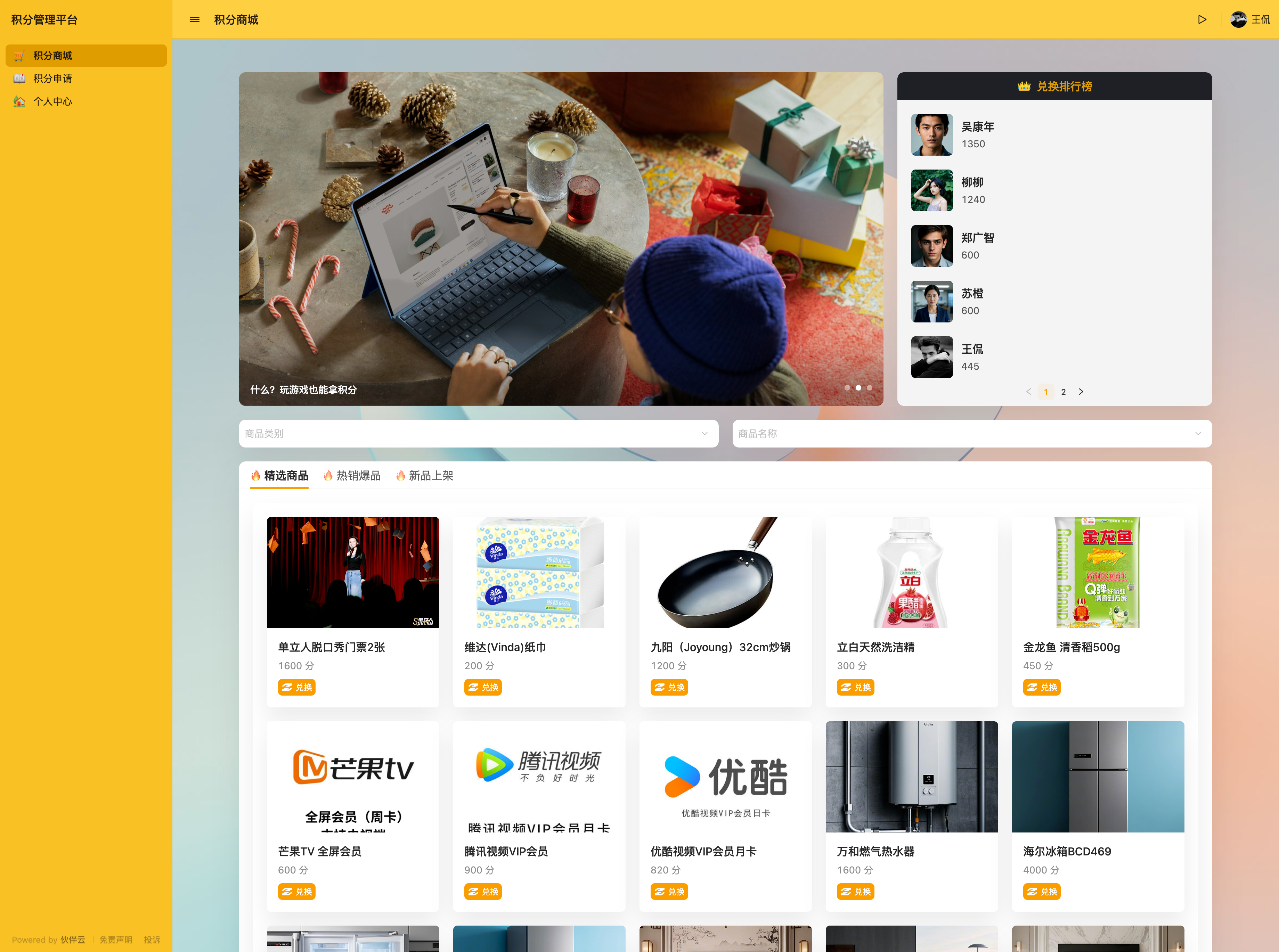
目录
欧盟医疗器械管理需遵守哪些具体要求?
欧盟医疗器械管理体系复杂完善,旨在确保产品安全、有效、质量可控。它以指令和法规为核心框架,按风险将器械分三类:一类低风险,制造商需满足基本质量管理要求并自我声明;二类中等风险,要合格评定;三类高风险,管理最严,需大量临床评估数据。质量管理涵盖全生命周期,上市前审批因类别而异,上市后仍持续监管。企业遵守规定至关重要,可避免处罚、树立形象等。
用户关注问题
欧盟医疗器械管理需遵守哪些主要法规?
比如说我是一家医疗器械公司,想把产品卖到欧盟去,但是我不太清楚那边对医疗器械管理都有啥法规要遵守,感觉一头雾水啊。
在欧盟,医疗器械管理需遵守多项重要法规。其中最关键的是医疗器械指令(MDD)和医疗器械法规(MDR)。MDD为传统的医疗器械管理框架,它按照风险等级将医疗器械分为不同类别,并规定了相应的合格评定程序等内容。而MDR是更新更全面的法规,其进一步加强了对医疗器械安全性、有效性的监管要求。例如,对于高风险医疗器械,要求更严格的临床试验和上市后监督等。这就要求企业在产品研发、生产、销售等各个环节都要严格遵循这些法规的要求,确保产品合规。如果您想深入了解如何让您的医疗器械顺利进入欧盟市场并完全符合法规要求,可以免费注册试用我们的咨询服务,我们会有专业团队为您详细解答。

欧盟医疗器械管理需遵守的认证流程是什么?
我是做医疗器械的,知道要进欧盟得遵守他们的管理规定,那具体认证流程咋样啊?就好比我要过一道道关卡一样,我都不知道先迈哪条腿。
首先,要确定您的医疗器械在欧盟分类体系中的类别,因为不同类别认证流程有区别。一般来说,低风险的一类医疗器械通常采用自我声明的方式,但也要满足基本的安全和性能要求并建立技术文件。对于二类及以上医疗器械,需要指定欧盟授权代表,并且要经过公告机构的审核。例如,二类医疗器械要进行产品测试、技术文件审查、质量管理体系审核等流程。然后由公告机构颁发CE认证标志,才能合法地在欧盟市场销售。整个过程复杂且严谨,需要企业投入足够的精力和资源。如果您希望更系统地掌握这个流程,欢迎预约演示我们的专业培训课程,帮助您轻松应对欧盟医疗器械认证流程。
欧盟医疗器械管理需遵守,对小型企业有何特殊要求?
我开了个小的医疗器械厂,想往欧盟卖点东西,我就想知道欧盟那边管医疗器械的时候,对像我这样的小企业有没有啥特别的要求呢?会不会因为我小就放松点,还是更严格啊?
欧盟医疗器械管理对小型企业并没有放松要求,但在执行上有一定灵活性。从优势(Strengths)方面看,小型企业往往更加灵活,能更快适应法规的一些新变化。然而,劣势(Weaknesses)在于它们资源有限,例如资金和人力方面难以承担复杂的法规遵从成本。机会(Opportunities)在于可以借助外部的专业服务机构来弥补自身不足。威胁(Threats)就是如果不能满足法规要求,很容易被市场淘汰。小型企业同样需要按照医疗器械的分类遵守相关法规,如建立完善的质量体系、进行必要的产品检测等。不过在某些情况下,例如提交技术文件等方面,可以与公告机构协商更适合自身规模的方案。如果您的小型企业正面临这些困扰,可点击免费注册试用我们为小企业量身定制的合规指导服务。
免责申明:本文内容通过 AI 工具匹配关键字智能整合而成,仅供参考,伙伴云不对内容的真实、准确、完整作任何形式的承诺。如有任何问题或意见,您可以通过联系 12345@huoban.com 进行反馈,伙伴云收到您的反馈后将及时处理并反馈。