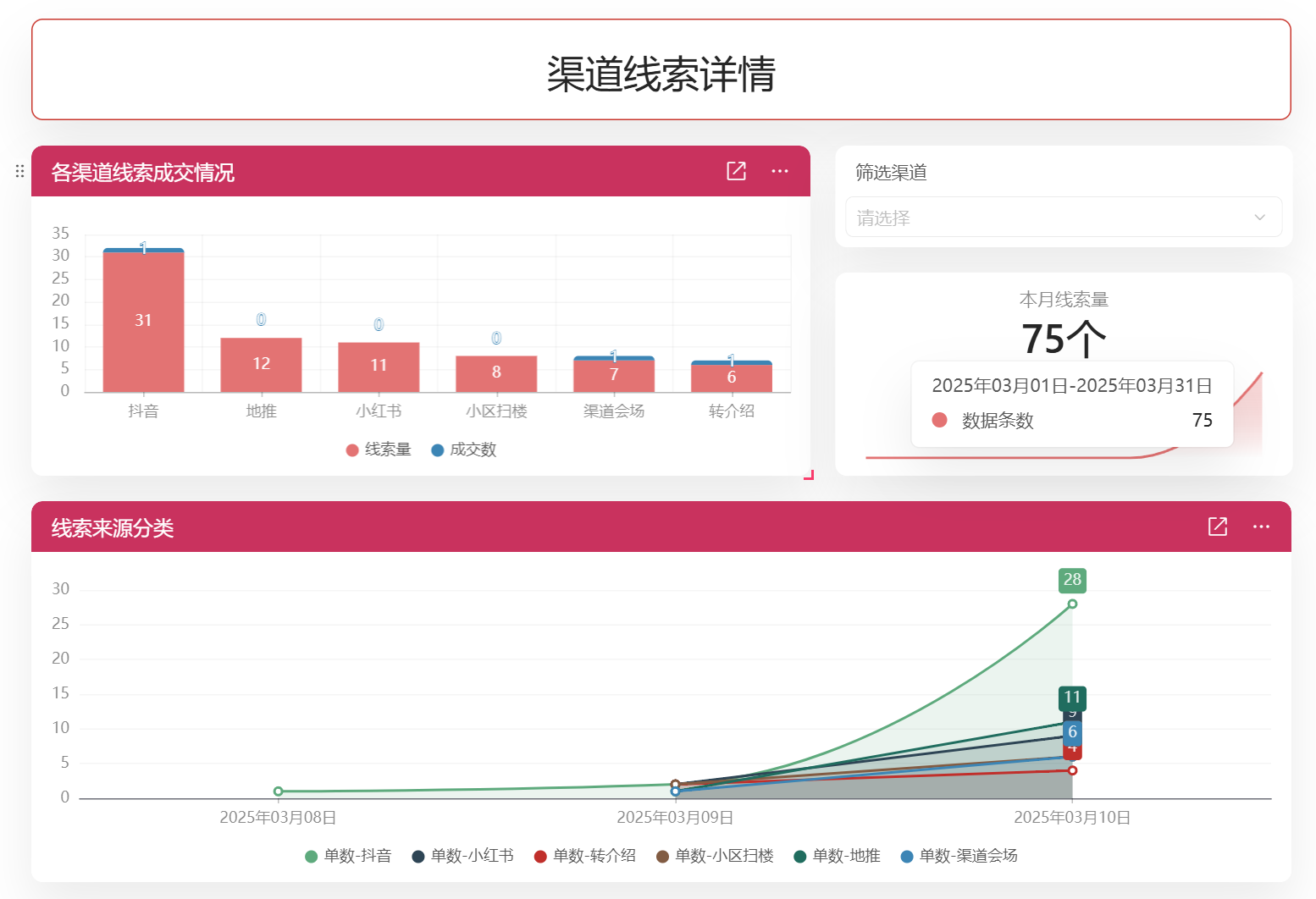
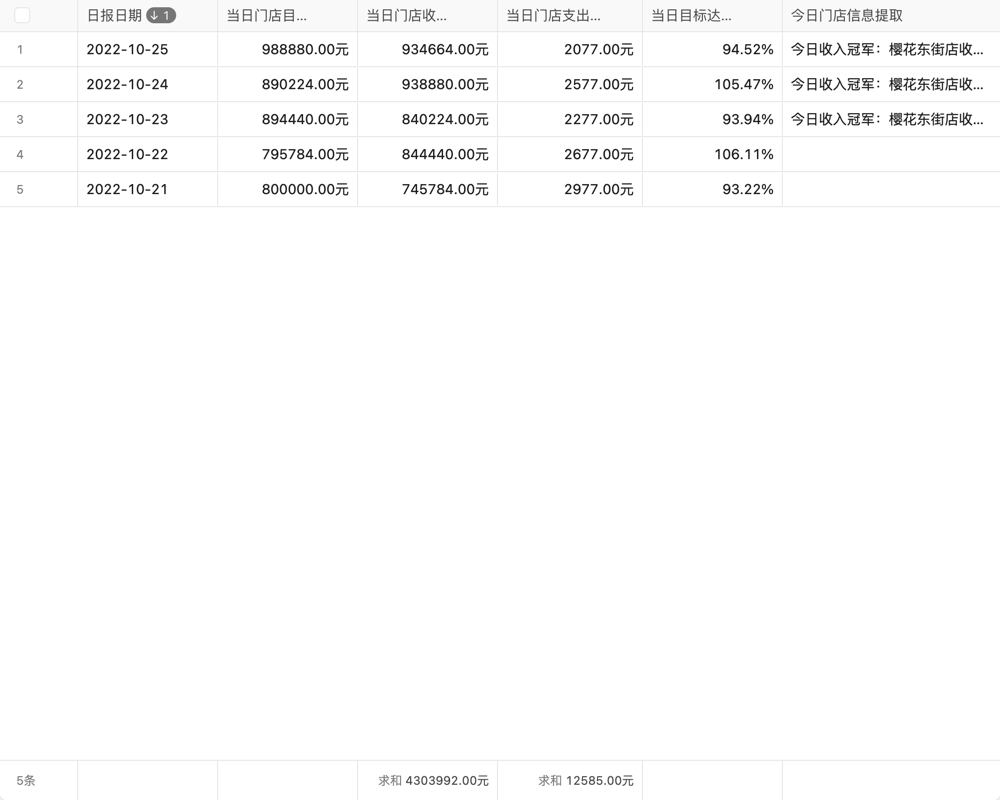
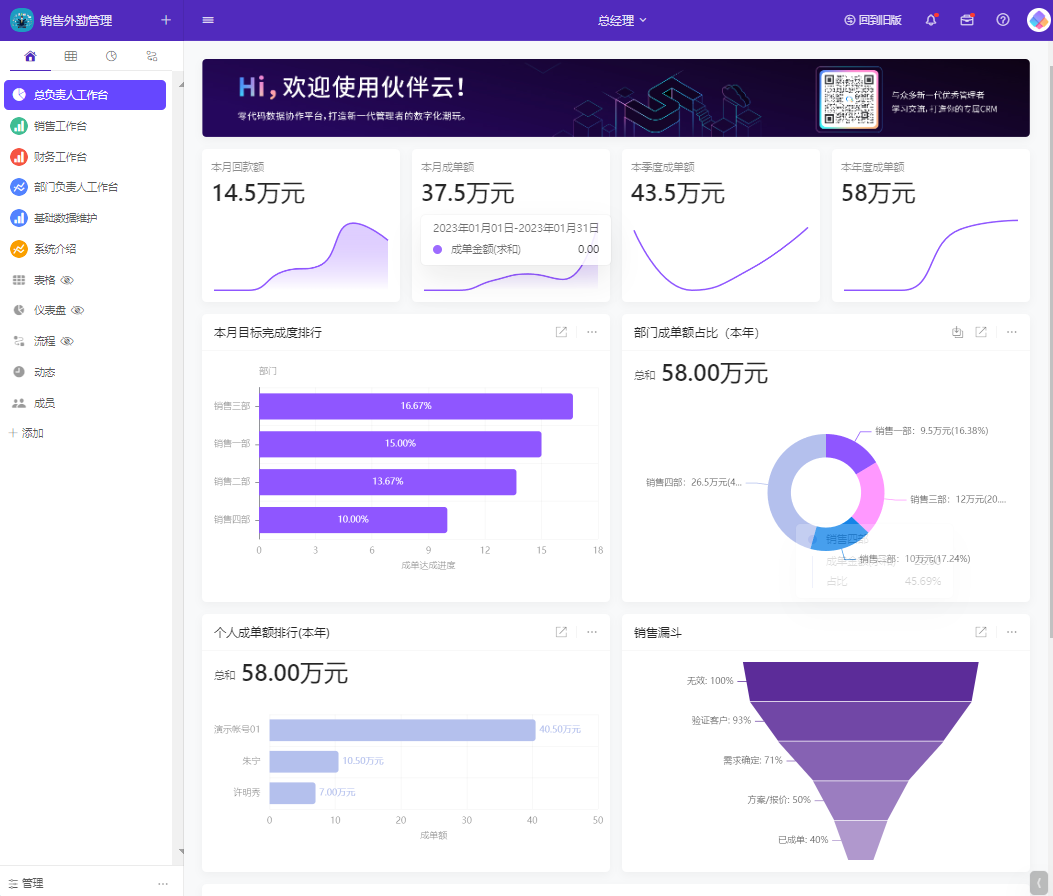
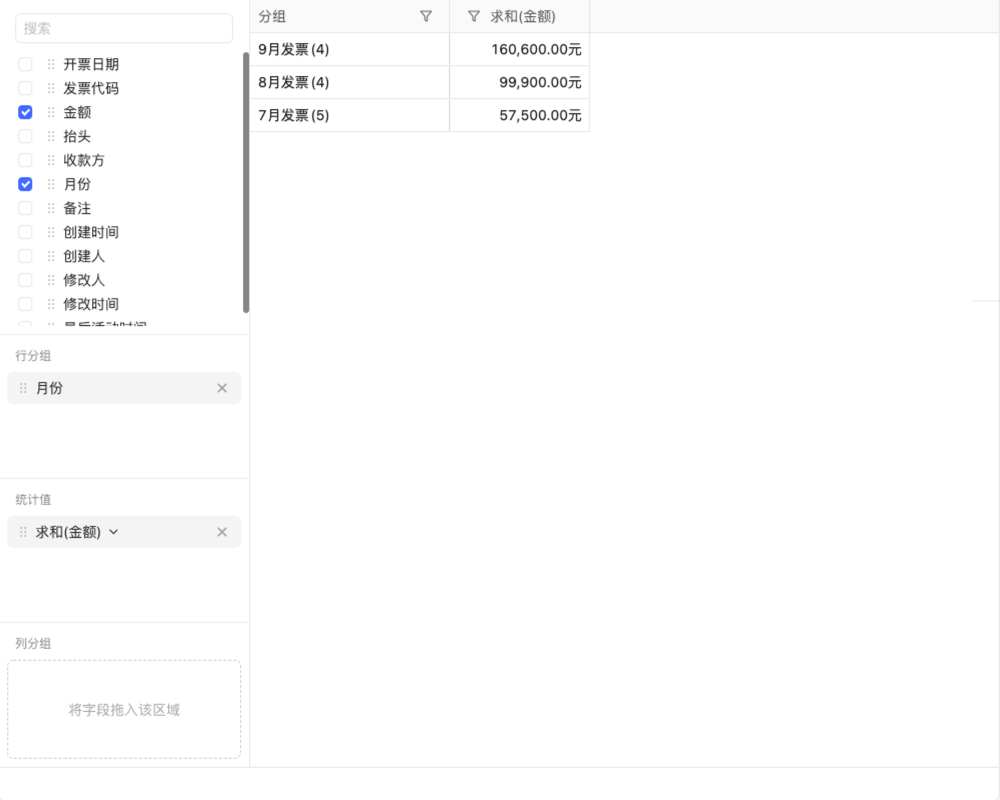
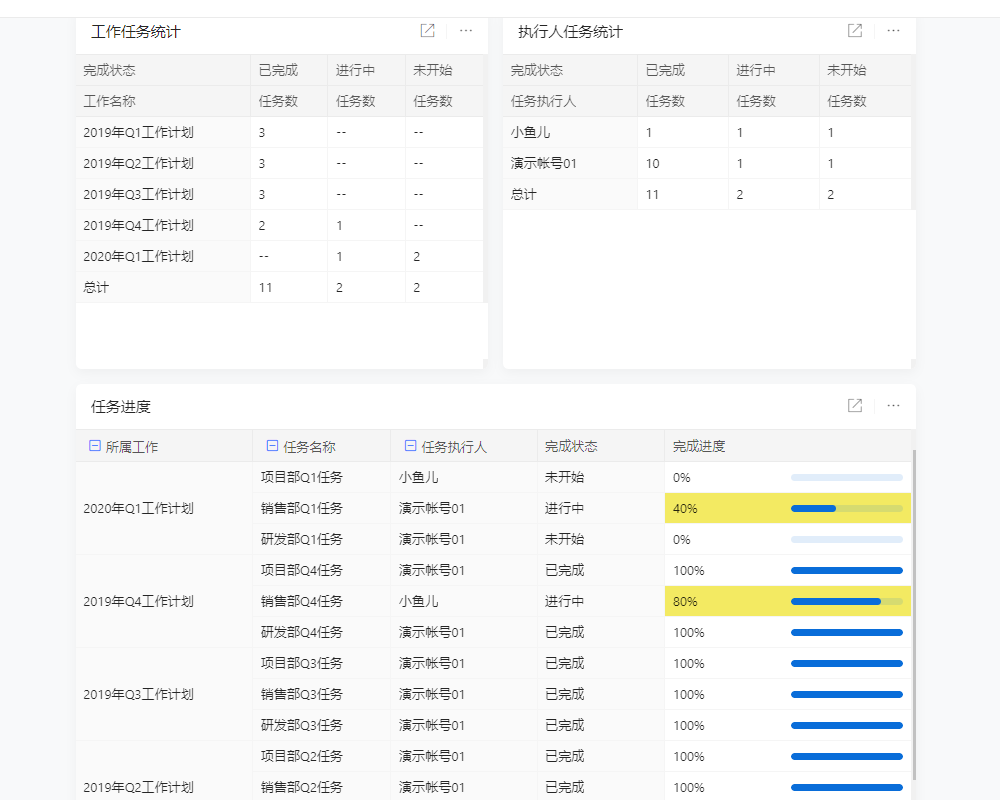
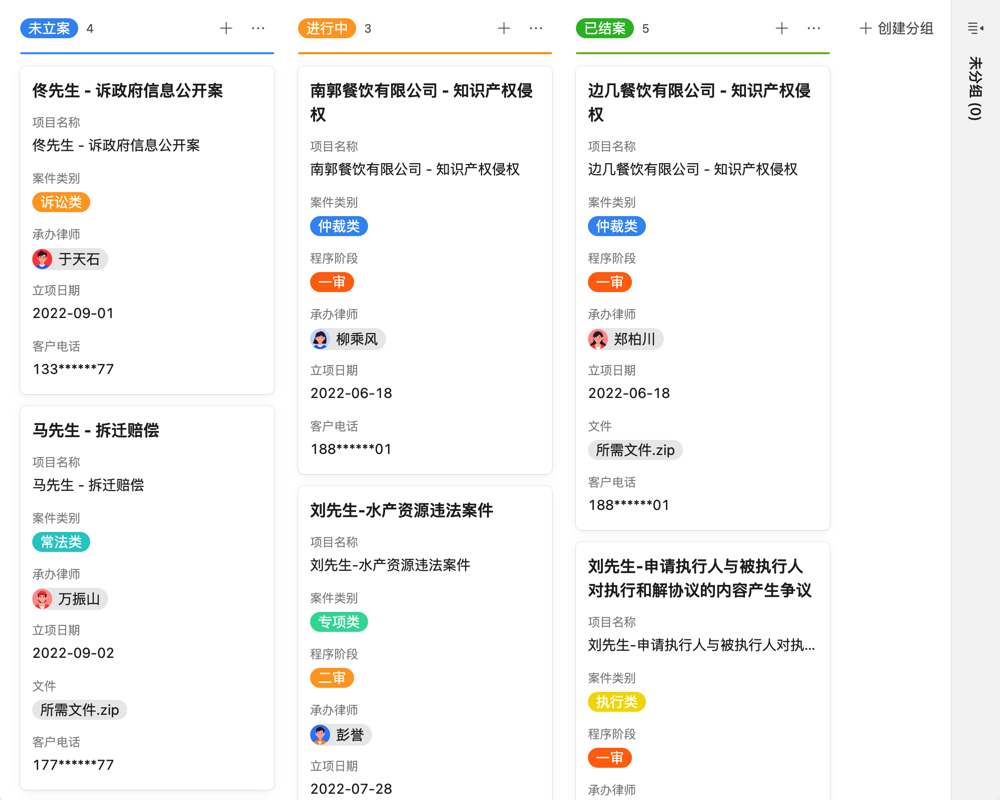
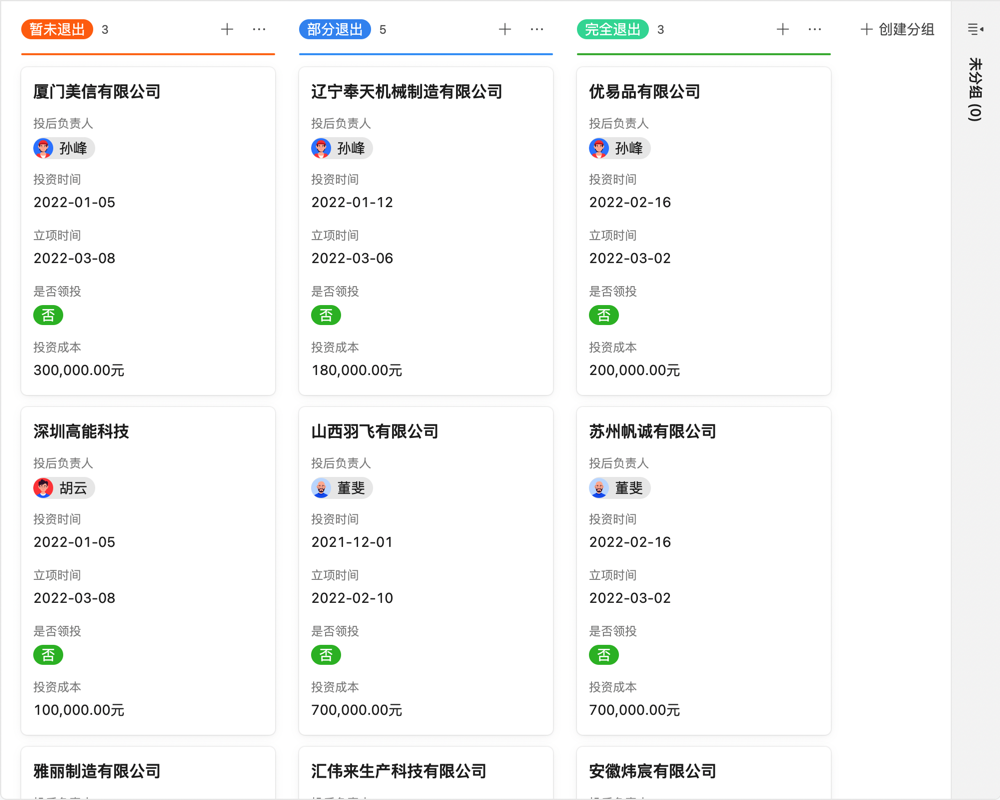
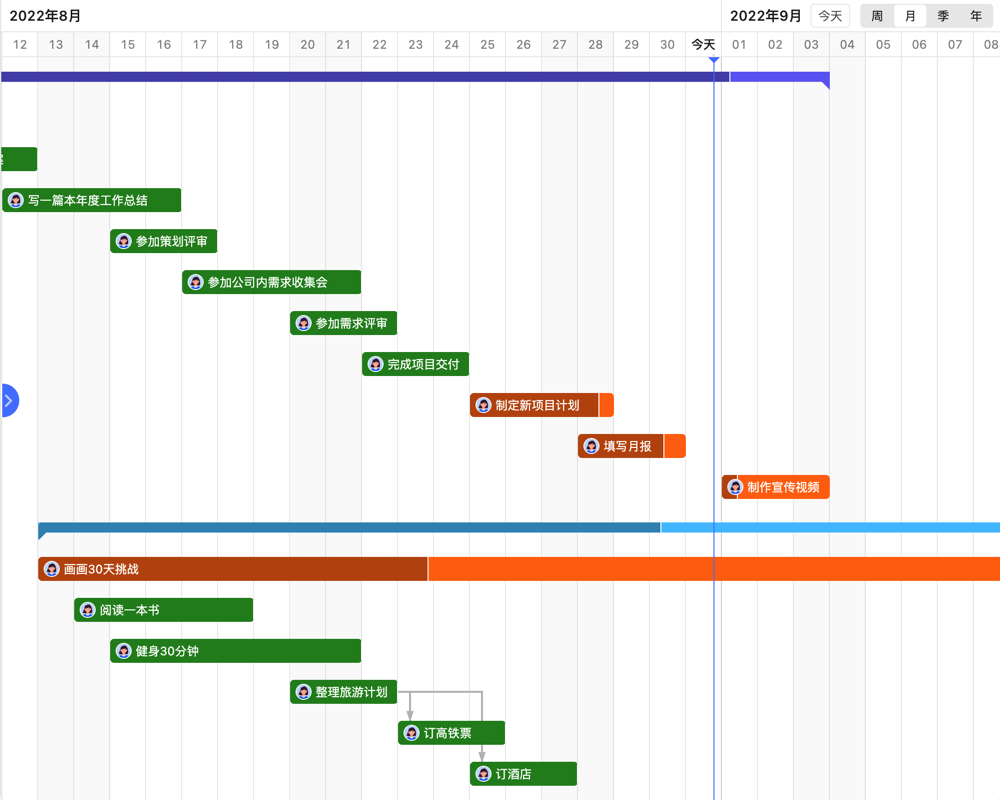
目录
如何全面解析欧洲医疗器械监督管理的严格要求?
欧洲医疗器械市场因其严格的监管体系备受瞩目。本文深入解析欧洲医疗器械监督管理的概览、监管体系、注册流程、新趋势及应对挑战的策略,帮助您更好地了解这一领域,确保医疗器械产品符合欧盟标准,顺利进入欧洲市场。文末还有惊喜等着您,免费注册试用或预约演示,让您的产品更快获得CE认证!
用户关注问题
欧洲医疗器械监督管理有哪些主要法规?
想了解在欧洲市场上销售医疗器械,需要遵守哪些主要的监督管理法规?这些法规对进口医疗器械有哪些具体要求?
欧洲医疗器械监督管理主要涉及《医疗器械指令》(MDD)、《有源植入性医疗器械指令》(AIMDD)以及后续的《医疗器械法规》(MDR)和《体外诊断医疗器械法规》(IVDR)等。这些法规对医疗器械的分类、注册、认证、生产、销售以及市场监管等方面做出了详细规定。对于进口医疗器械,需满足欧盟的安全性和有效性要求,通过相应的CE认证,并遵循欧盟的监管流程。此外,还需注意欧盟不断更新的技术标准和市场准入要求。若您想深入了解这些法规的具体内容和实施细节,不妨点击免费注册试用我们的法规数据库,获取最新的欧洲医疗器械监督管理信息。

如何确保欧洲市场医疗器械的合规性?
想在欧洲市场销售医疗器械,但不知道该如何确保产品的合规性,避免违规风险?
确保欧洲市场医疗器械的合规性,需要从多个方面入手:1. 了解并遵守相关法规;2. 进行充分的产品测试和认证;3. 建立完善的质量管理体系;4. 加强市场监督和风险管理。具体而言,您需要关注欧盟的医疗器械法规更新,确保产品符合最新的安全性和有效性要求;通过CE认证等程序,证明产品的合规性;同时,建立有效的质量管理体系,确保生产、销售和售后服务的全过程可追溯。此外,还需定期评估市场风险,及时采取措施应对。我们提供专业的合规咨询和认证服务,助您轻松应对欧洲市场的合规挑战,欢迎预约演示了解详情。
欧洲医疗器械注册流程是怎样的?
想知道在欧洲注册医疗器械需要经过哪些步骤?需要准备哪些材料和文件?
欧洲医疗器械注册流程一般包括以下几个步骤:1. 确定产品分类;2. 准备注册材料;3. 提交注册申请;4. 进行技术评估和审核;5. 获得注册证书。在准备注册材料时,您需要提供产品的技术文档、性能测试报告、质量管理体系文件等。提交申请后,欧盟相关机构将对您的产品进行技术评估和审核,确保符合安全性和有效性要求。一旦通过审核,您将获得注册证书,允许在欧洲市场销售产品。我们拥有丰富的注册经验和专业团队,可为您提供一站式注册服务,点击免费咨询了解更多注册流程。
欧洲医疗器械监督管理对市场的影响有哪些?
欧洲医疗器械监督管理的加强,对市场有哪些具体影响?对医疗器械企业有哪些挑战和机遇?
欧洲医疗器械监督管理的加强,对市场产生了深远影响。一方面,它提高了医疗器械的安全性和有效性要求,保障了患者的权益,提升了公众对医疗器械的信任度。另一方面,这也对医疗器械企业带来了挑战,如增加了注册和认证成本、提高了产品质量要求等。然而,这也为企业带来了机遇,如推动技术创新、提升品牌影响力等。因此,企业需要积极应对监管变化,加强合规管理,提升产品质量和服务水平。我们提供专业的合规咨询和培训服务,助您把握市场机遇,应对监管挑战,欢迎预约演示了解我们的服务。
免责申明:本文内容通过 AI 工具匹配关键字智能整合而成,仅供参考,伙伴云不对内容的真实、准确、完整作任何形式的承诺。如有任何问题或意见,您可以通过联系 12345@huoban.com 进行反馈,伙伴云收到您的反馈后将及时处理并反馈。