目录
医疗器械的样机管理办法真的那么重要吗?全面解析与实施建议
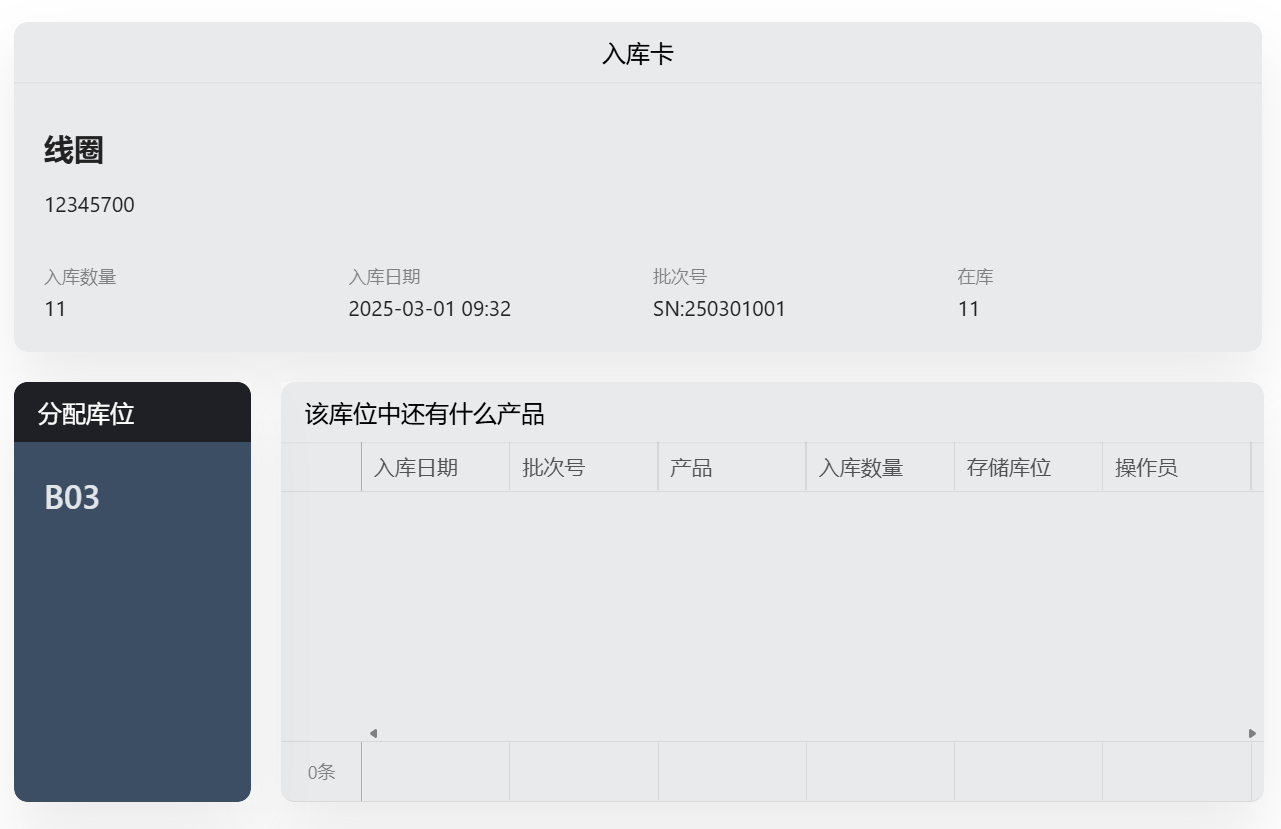
在医疗器械研发中,样机管理办法是确保产品顺利推进的关键。本文详解样机编号、存储、测试及版本控制等核心内容,结合实际案例,助您高效实施!了解规范化的管理如何降低风险、提升效率,为您的项目保驾护航。
用户关注问题
医疗器械样机管理办法的核心内容是什么?
比如,我是一名医疗器械企业的研发人员,想知道国家对样机管理有哪些具体规定?这些规定对我们日常研发工作有什么影响?
医疗器械样机管理办法的核心内容主要围绕样机的研发、测试、验证以及合规性展开。以下是关键点:
1. 样机的研发阶段:
需要确保设计符合相关法规和标准,例如ISO 13485等质量管理体系要求。
2. 样机的测试与验证:
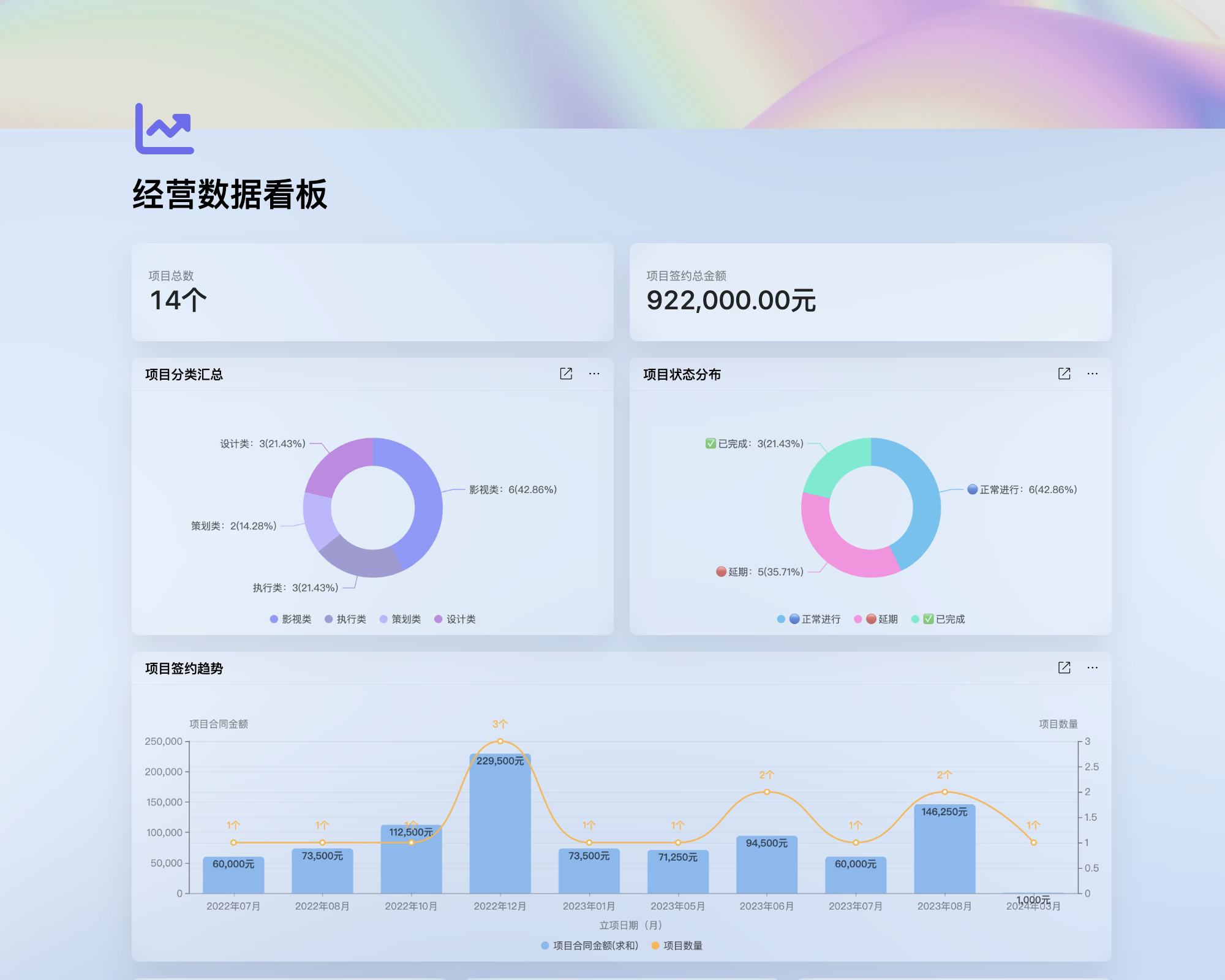
必须经过严格的性能测试、安全评估和临床前研究,确保其在实际使用中的可靠性和安全性。
3. 合规性要求:
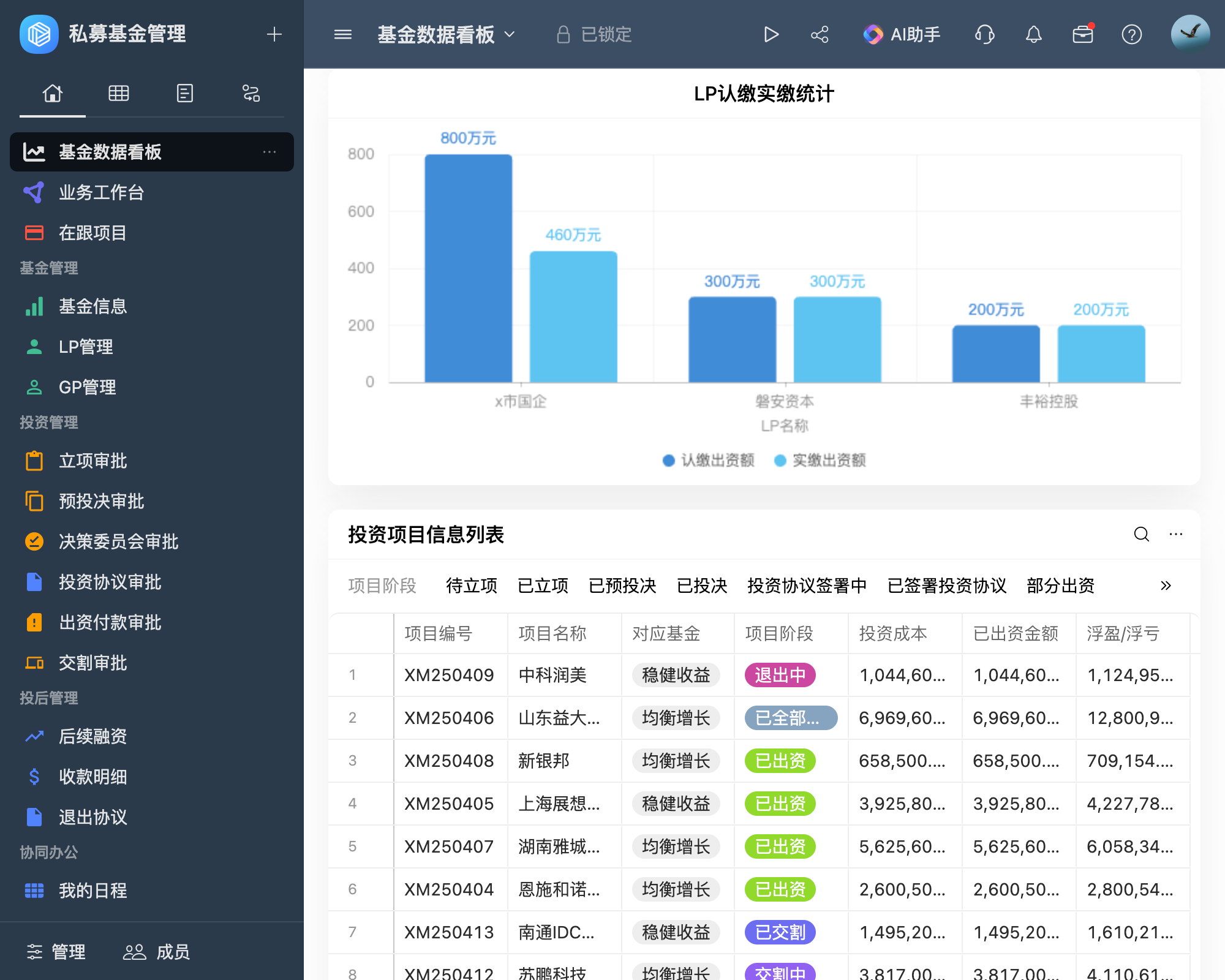
样机需满足NMPA(国家药品监督管理局)或其他国际监管机构的注册申报要求,确保从研发到上市的全流程可追溯。
建议您根据自身企业的需求,注册试用我们的平台,获取更多关于样机管理的专业指导和模板。

医疗器械样机管理办法对企业有哪些具体要求?
假如我是企业负责人,想知道我们公司需要在样机管理方面做哪些准备,才能符合国家的规定?
医疗器械样机管理办法对企业的要求主要体现在以下几个方面:
1. 质量管理体系:
企业需建立完善的质量管理体系,涵盖样机的设计开发、生产控制和风险管理。
2. 文件记录管理:
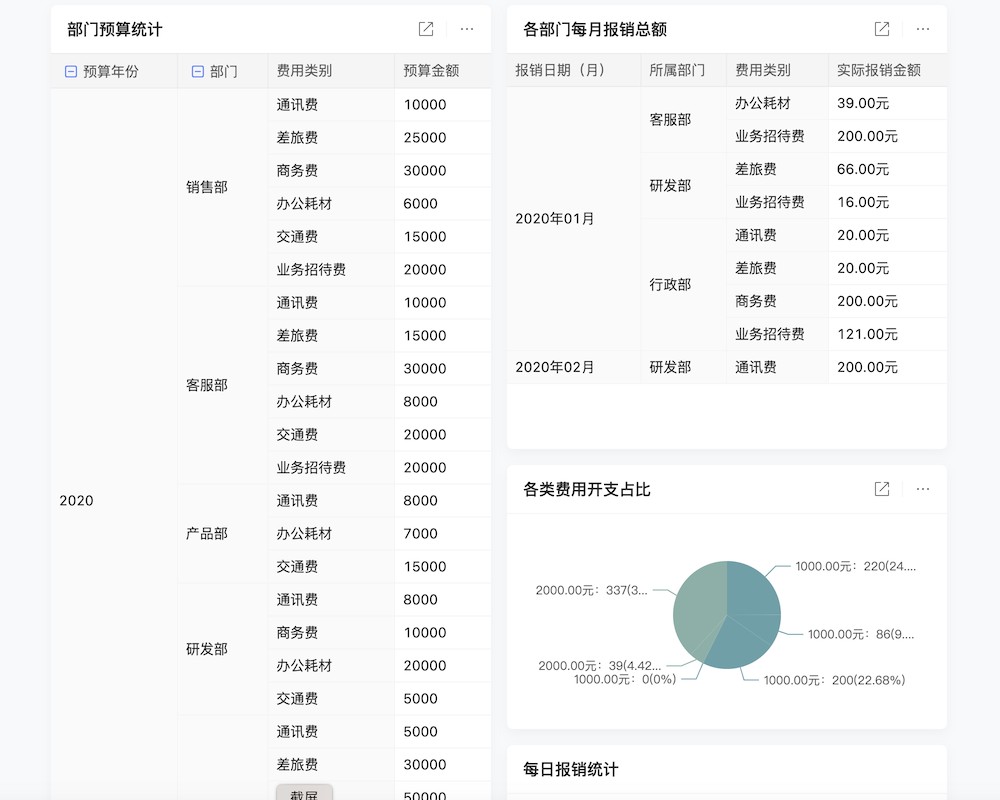
所有样机相关的技术文件、实验数据和测试报告都需完整保存,并定期更新。
3. 风险评估与控制:
企业应识别样机可能存在的风险,并采取措施降低或消除这些风险。
通过SWOT分析可以看出,企业如果能提前做好这些准备,不仅可以降低合规风险,还能提升市场竞争力。如果您想了解更多详细信息,欢迎预约演示我们的解决方案。
如何制定医疗器械样机的管理流程?
作为一名质量管理经理,我想知道制定一个科学合理的样机管理流程应该从哪些方面入手?
制定医疗器械样机的管理流程可以按照以下步骤进行:
1. 明确目标:
确定样机管理的目标,例如提高研发效率、降低失败率等。
2. 设计开发阶段:
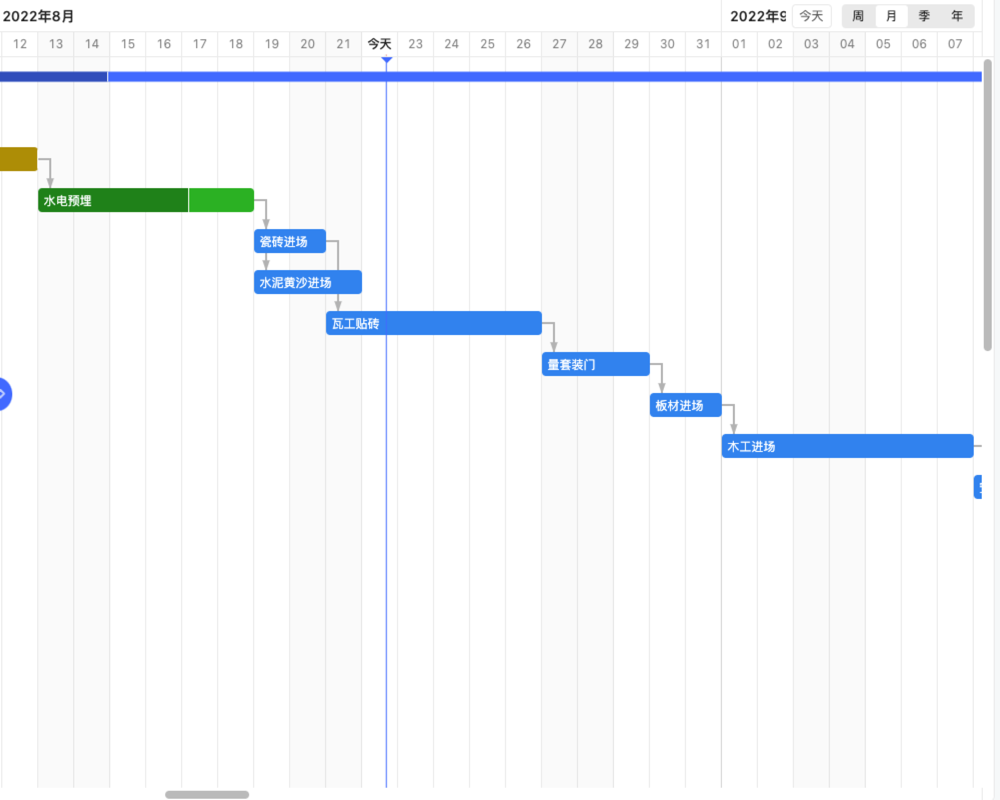
包括需求分析、设计输入、设计输出和设计验证等内容。
3. 测试与验证阶段:
进行功能性、安全性测试,并记录所有数据。
4. 文件归档与评审:
整理所有相关文件,并组织内部评审。
为了帮助您更好地制定流程,我们提供了一系列标准化模板和工具,您可以免费注册试用,体验我们的服务。
免责申明:本文内容通过 AI 工具匹配关键字智能整合而成,仅供参考,伙伴云不对内容的真实、准确、完整作任何形式的承诺。如有任何问题或意见,您可以通过联系 12345@huoban.com 进行反馈,伙伴云收到您的反馈后将及时处理并反馈。