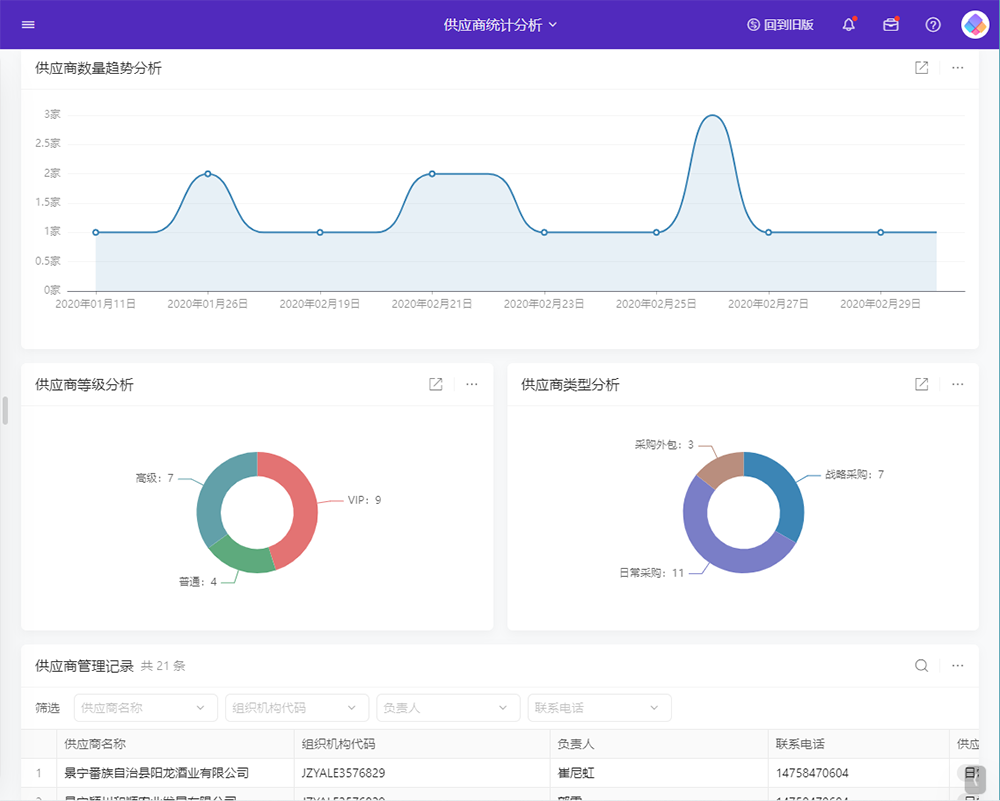
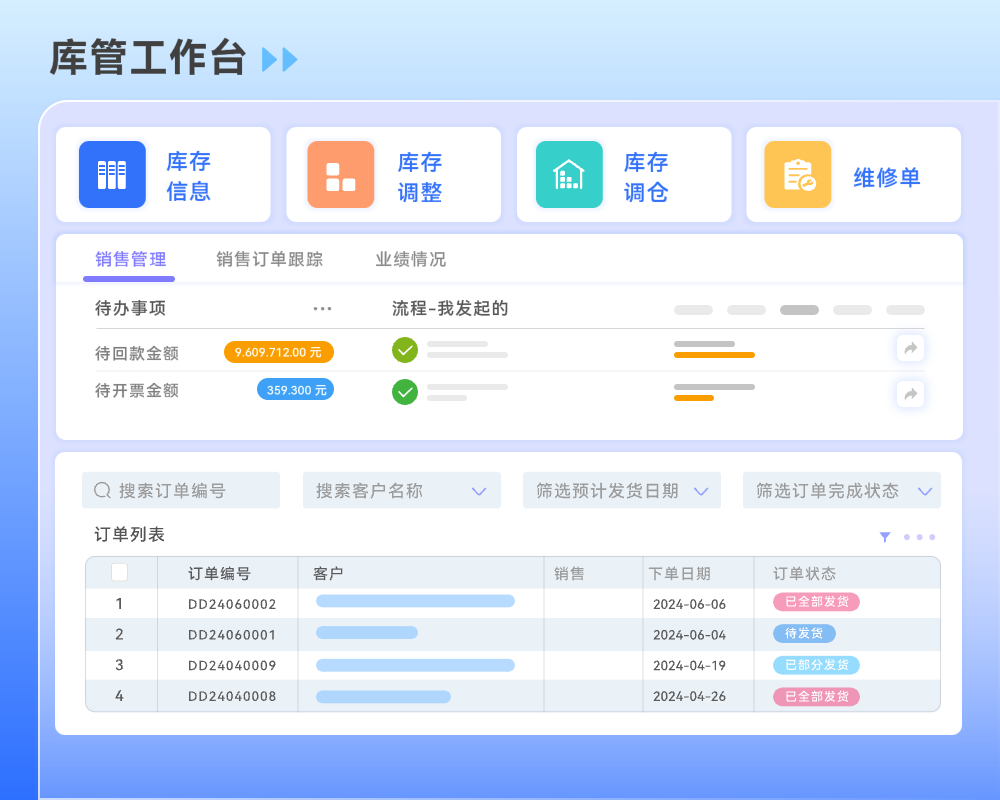
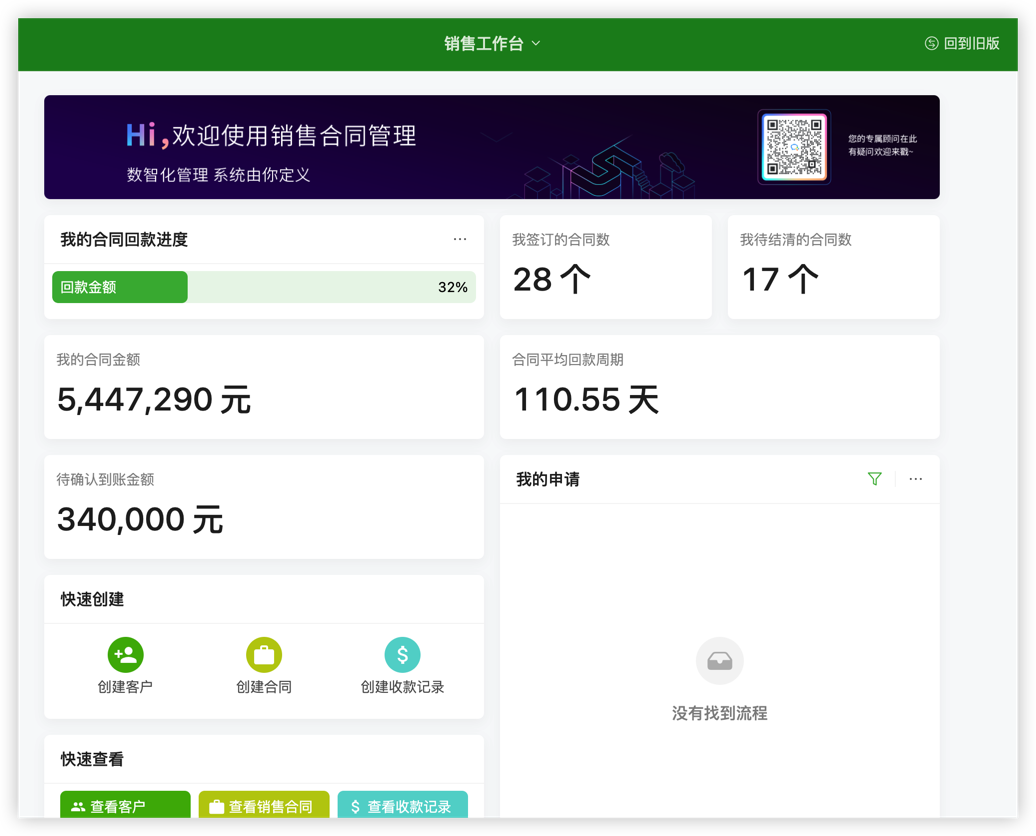
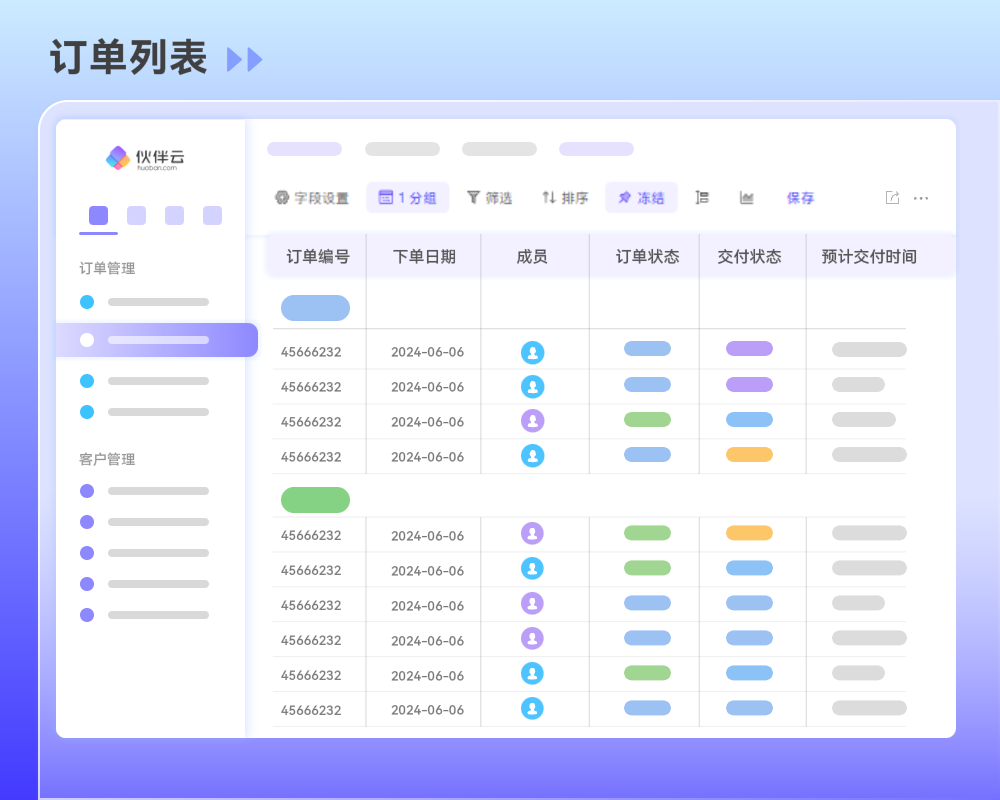
目录
《美国非处方医疗器械管理办法:您了解多少?》
在美国,非处方医疗器械市场规模渐增,其管理办法十分重要。按风险等级可分为低、中、高风险类,按功能用途分诊断、治疗、预防类。审批流程含申报前准备、向FDA提交申请、审查评估及根据审批结果行动等。标签和说明书有严格规定,在生产方面有设施环境、原材料采购检验、生产过程质量监控等要求。FDA负责市场监管且有不良事件监测系统。
用户关注问题
美国非处方医疗器械管理办法有哪些要点?
比如说我想在美国卖一些非处方的医疗器械,像体温计之类的,那我得知道那边对这些东西管理都有啥要求啊,这管理办法里重点都是啥呢?
美国非处方医疗器械管理办法有几个关键要点。首先,分类管理很重要,不同类型的非处方医疗器械根据风险程度被分入不同类别。例如低风险的如普通绷带等,管理相对宽松;而像一些简易的家用检测设备风险稍高些的,则有更多的质量和标识要求。其次是标识要求,产品包装上必须清晰标明使用方法、适用范围、警告事项等内容。再者,制造商要遵循质量管理体系的规定确保产品质量稳定可靠。如果您想深入了解美国非处方医疗器械管理的详细情况,可以点击免费注册试用我们的资讯服务,获取更多专业解读。

如何遵守美国非处方医疗器械管理办法进行产品注册?
我有个非处方医疗器械想在美国市场卖,听说要注册,可按照这个管理办法咋注册呢?感觉一头雾水啊。
遵守美国非处方医疗器械管理办法进行产品注册主要有以下步骤。第一步,确定产品分类,根据产品的特性、用途和风险程度找到对应的分类。第二步,准备相关文件,包括产品说明书(里面详细说明使用、注意事项等)、技术文件(例如产品设计、制造流程等)等。第三步,向美国食品药品监督管理局(FDA)提交注册申请。第四步,等待审核,审核过程中可能会要求补充材料或者对产品进行检测等。这里需要提醒的是,整个过程较为复杂,为了确保顺利注册,您可以预约演示我们的专业指导服务,帮助您更好地应对。从SWOT分析来看,优势在于如果注册成功就能进入广阔的美国市场;劣势是注册流程复杂耗费时间精力;机会是美国市场需求大;威胁则是竞争激烈且法规随时可能变化。
美国非处方医疗器械管理办法对进口产品有特殊要求吗?
我的非处方医疗器械是从国外生产然后想卖到美国的,就想知道这个管理办法对进口的这种有没有额外的规定呀?
美国非处方医疗器械管理办法对进口产品确实有特殊要求。进口产品首先要符合一般的非处方医疗器械管理规定,如上述提到的分类管理、标识要求等。此外,进口商需要承担一定的责任,确保进口的产品合规。比如要保留进口产品的相关记录以便追溯,要确保产品在运输和存储过程中符合质量要求等。对于外国制造商,可能还需要指定美国代理人来处理相关事务。如果您想了解如何确保您的进口产品完全符合要求,欢迎点击免费注册试用我们的合规检查工具。从象限分析来看,根据产品来源国和产品类型的不同,其面临的合规难度处于不同象限,例如来自监管体系相似国家的简单产品可能较易合规,而来自监管差异大国家的复杂产品则面临较大挑战。
免责申明:本文内容通过 AI 工具匹配关键字智能整合而成,仅供参考,伙伴云不对内容的真实、准确、完整作任何形式的承诺。如有任何问题或意见,您可以通过联系 12345@huoban.com 进行反馈,伙伴云收到您的反馈后将及时处理并反馈。