目录
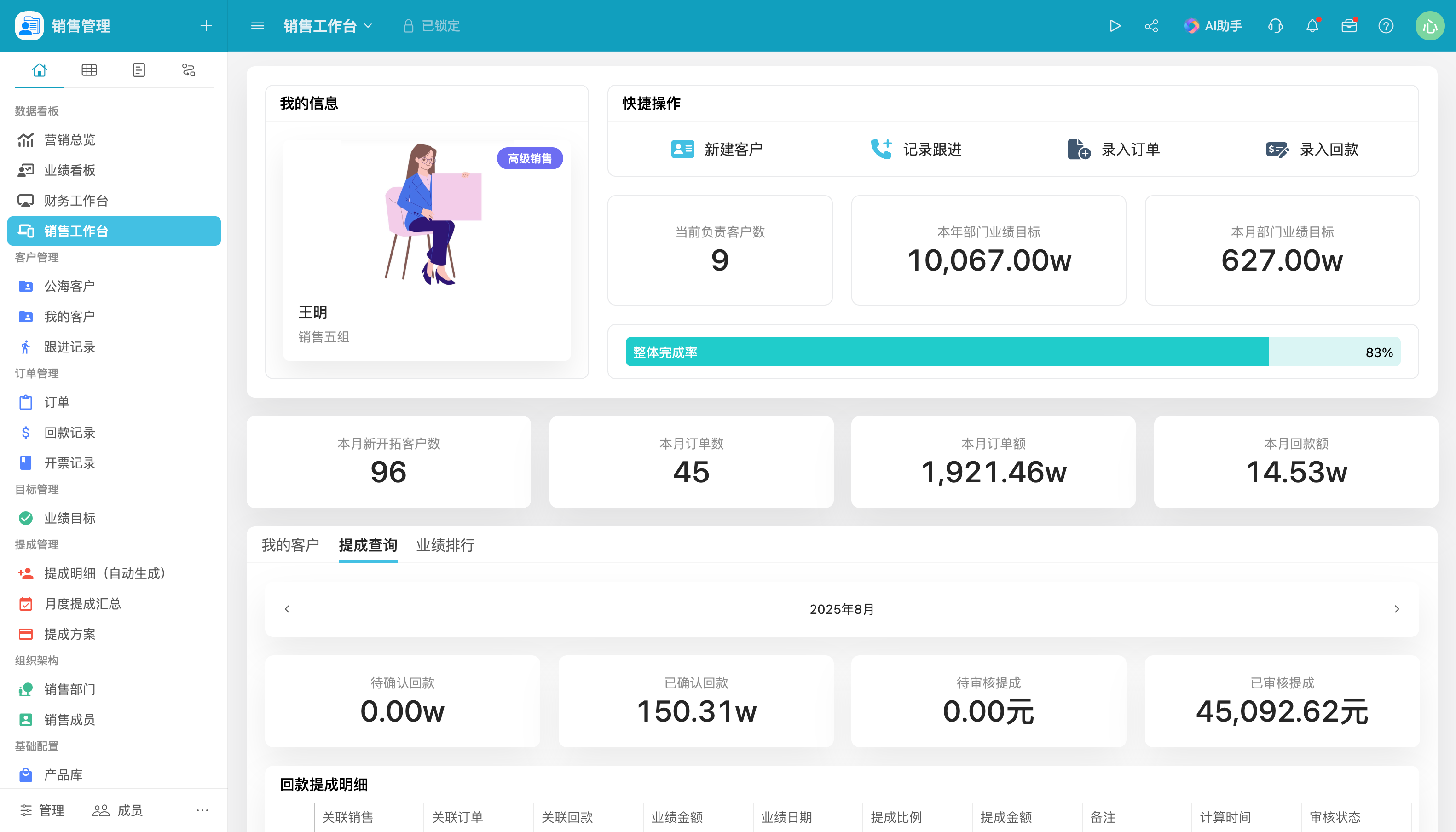
医疗器械欧盟风险管理为何如此关键?
了解医疗器械欧盟风险管理的重要性是进入欧盟市场的关键一步。本文深入探讨了风险管理的核心步骤、MDR 2017/745法规要求以及实际应用案例,帮助企业更好地应对挑战并优化流程。通过数字化工具和跨职能团队协作,您可以确保产品在整个生命周期内符合欧盟标准,同时保障患者安全。
用户关注问题
医疗器械在欧盟市场需要遵循哪些风险管理要求?
假如您是一家医疗器械制造商,正准备将产品出口到欧盟市场,但对欧盟的风险管理要求还不太了解。您可能会想问:我的产品需要满足哪些具体的风险管理规定才能顺利进入欧盟市场呢?
在欧盟市场,医疗器械风险管理主要依据MDD(Medical Device Directive)或MDR(Medical Device Regulation)的规定。以下是关键步骤和要求:
- 风险分析与评估:通过识别潜在危害,评估其可能性和严重性,并记录在风险管理计划中。
- 控制措施实施:采取设计改进、防护措施或提供使用说明等手段降低风险。
- 风险可接受性判断:根据欧盟标准(如EN ISO 14971),确定剩余风险是否可接受。
- 持续监控与改进:上市后需定期收集反馈,更新风险管理文档。
为确保您的产品完全合规,建议注册试用专业的医疗器械风险管理软件,它能帮助您高效完成这些步骤并生成符合要求的文档。

医疗器械企业如何应对欧盟MDR中的风险管理挑战?
如果您是医疗器械企业的负责人,可能正在苦恼如何应对欧盟MDR带来的更高风险管理要求。比如:我们该如何调整现有的流程来满足新的法规呢?
面对MDR的挑战,可以从以下几个方面入手:
- 加强团队培训:确保所有相关人员理解MDR的风险管理条款。
- 优化内部流程:重新审视现有流程,确保涵盖从设计开发到生产全过程的风险管理。
- 采用先进工具:引入专业软件辅助进行风险评估、跟踪和报告。
- 外部资源支持:考虑与经验丰富的咨询公司合作,获取专业指导。
通过以上措施,您可以更好地适应MDR的要求。同时,欢迎您预约演示我们的解决方案,了解更多实用功能。
欧盟医疗器械风险管理文档需要包括哪些内容?
作为质量管理人员,您可能正在准备一份完整的风险管理文档,但不确定是否涵盖了所有必要的内容。那么,一份合格的欧盟医疗器械风险管理文档到底应该包含什么呢?
一份完整的风险管理文档通常包括以下内容:
| 部分 | 具体内容 |
|---|---|
| 1. 风险管理计划 | 描述风险管理活动的目标、范围、职责分配及时间安排。 |
| 2. 风险分析报告 | 列出所有已识别的危害及其相关风险。 |
| 3. 风险评估结果 | 详细说明每个风险的定性和定量评估结果。 |
| 4. 风险控制措施 | 记录采取的降低风险的措施及其有效性验证。 |
| 5. 残余风险评价 | 分析剩余风险是否可接受。 |
| 6. 风险管理报告 | 总结整个风险管理过程及其结论。 |
如果您希望获得更系统化的支持,不妨尝试我们的在线平台,它能够协助生成规范的风险管理文档。
免责申明:本文内容通过 AI 工具匹配关键字智能整合而成,仅供参考,伙伴云不对内容的真实、准确、完整作任何形式的承诺。如有任何问题或意见,您可以通过联系 12345@huoban.com 进行反馈,伙伴云收到您的反馈后将及时处理并反馈。