目录
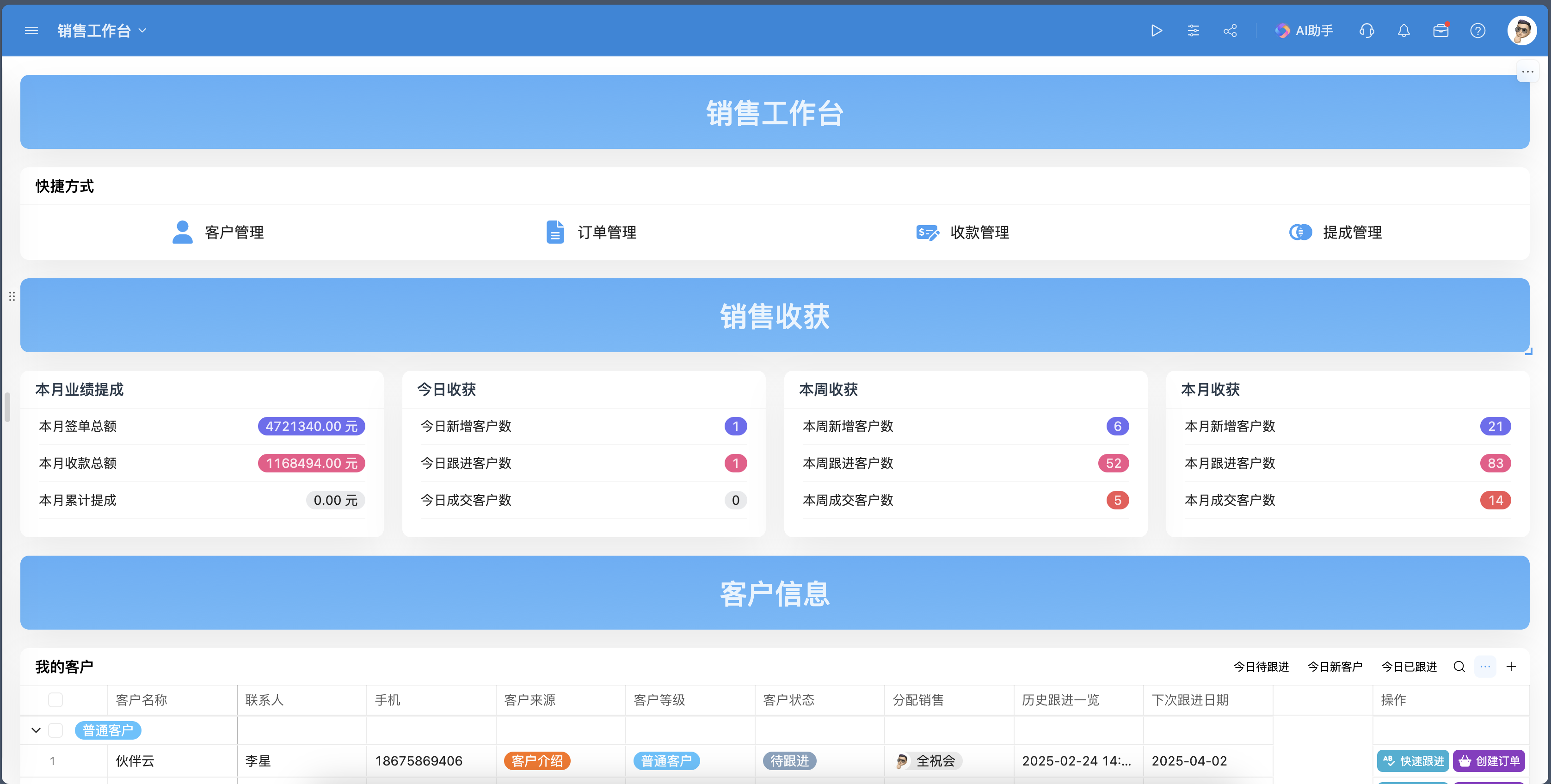
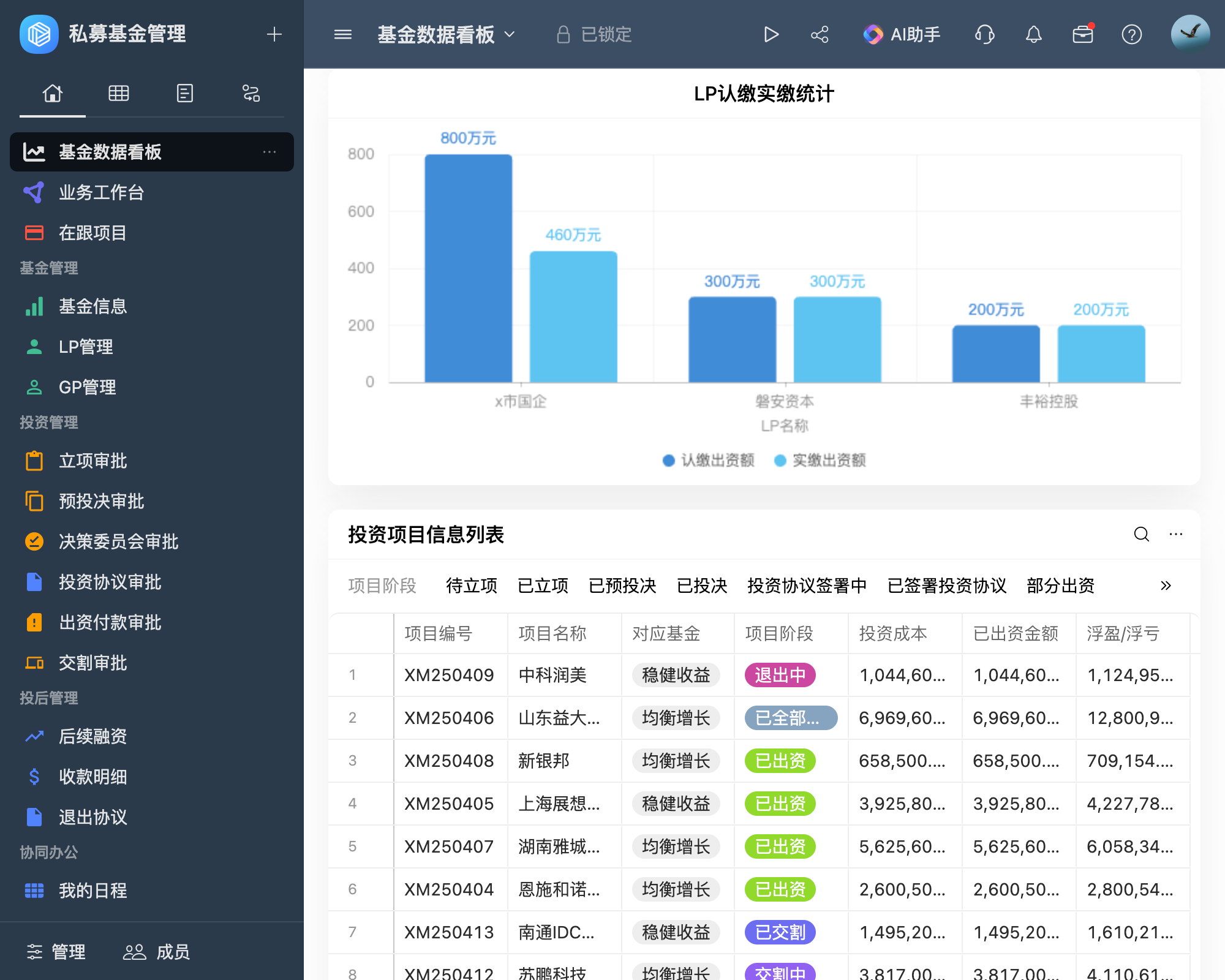
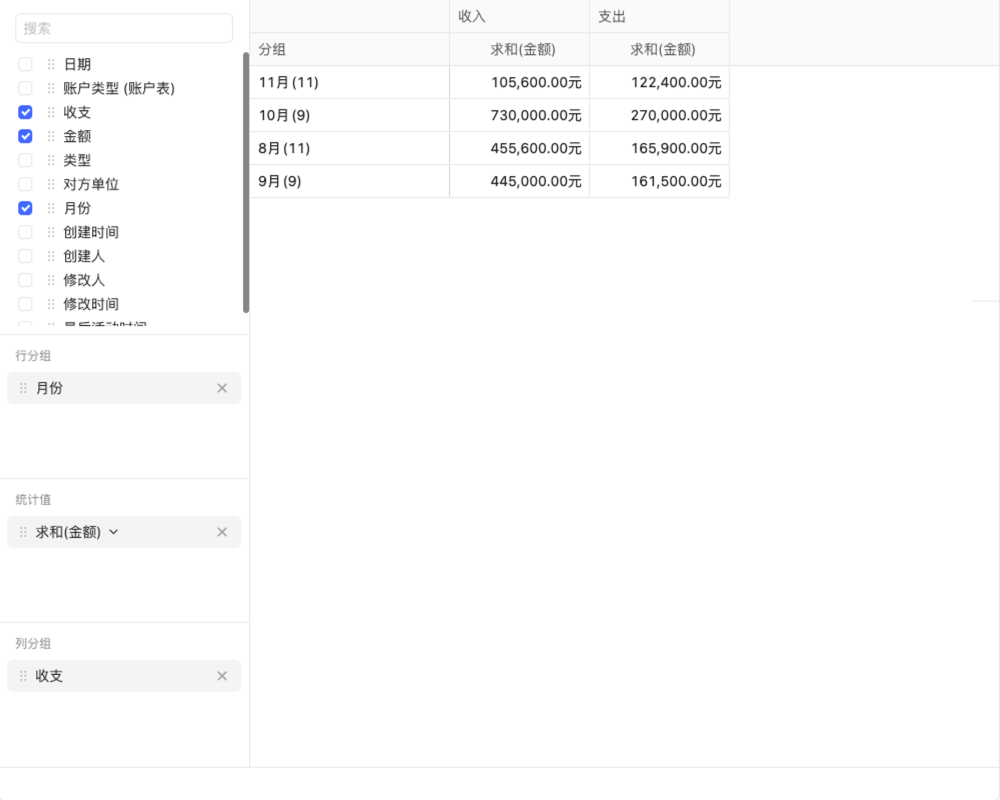
如何全面了解并实施最新的医疗器械产品管理办法?
医疗器械产品管理办法涵盖了研发、生产、流通、使用及后续监管等多个环节,确保医疗器械的安全性、有效性和可追溯性。本文详细介绍了医疗器械产品的分类管理、研发与生产、流通与使用等关键环节的管理要求,以及企业如何应对这些管理办法,提升产品质量和安全性。
用户关注问题
医疗器械产品管理办法具体包括哪些内容?
嘿,我听说医疗器械产品的管理有一套严格的规定,这管理办法里到底都说了些啥?比如怎么注册、怎么监管之类的。
医疗器械产品管理办法涵盖了从产品研发、生产、销售到使用的全生命周期管理。具体来说,它包括了医疗器械的分类管理、注册与备案流程、生产质量管理规范、经营与使用要求以及不良事件监测与处置等多个方面。比如,对于生产企业,需要遵循GMP(良好生产规范)标准,确保产品质量;对于经营者,则需取得相应的经营许可证,并严格遵守销售规定。此外,管理办法还强调了监管部门的职责与权限,以保障公众用械安全。想要深入了解这些规定吗?不妨点击我们的平台免费注册试用,获取更多专业解读哦!

如何合规地进行医疗器械产品的注册?
我想把自家研发的医疗器械推向市场,但听说注册流程挺复杂的,得怎么合规操作呢?
合规进行医疗器械产品注册,确实需要遵循一系列严格的步骤。首先,你需要明确产品的分类,根据风险等级选择合适的注册路径。接着,准备完整的注册资料,包括产品技术要求、检验报告、临床试验数据等。在提交注册申请前,务必确保所有资料的真实性与完整性。注册过程中,还需与监管部门保持密切沟通,及时响应补充资料或现场核查等要求。一旦注册成功,你将获得产品上市销售的合法资格。想要更顺利地完成注册流程吗?不妨预约我们的专业演示,获取一对一的指导服务吧!
医疗器械产品管理办法对生产企业有哪些具体要求?
作为医疗器械生产企业,我们得遵守哪些管理规定才能确保合规生产呢?
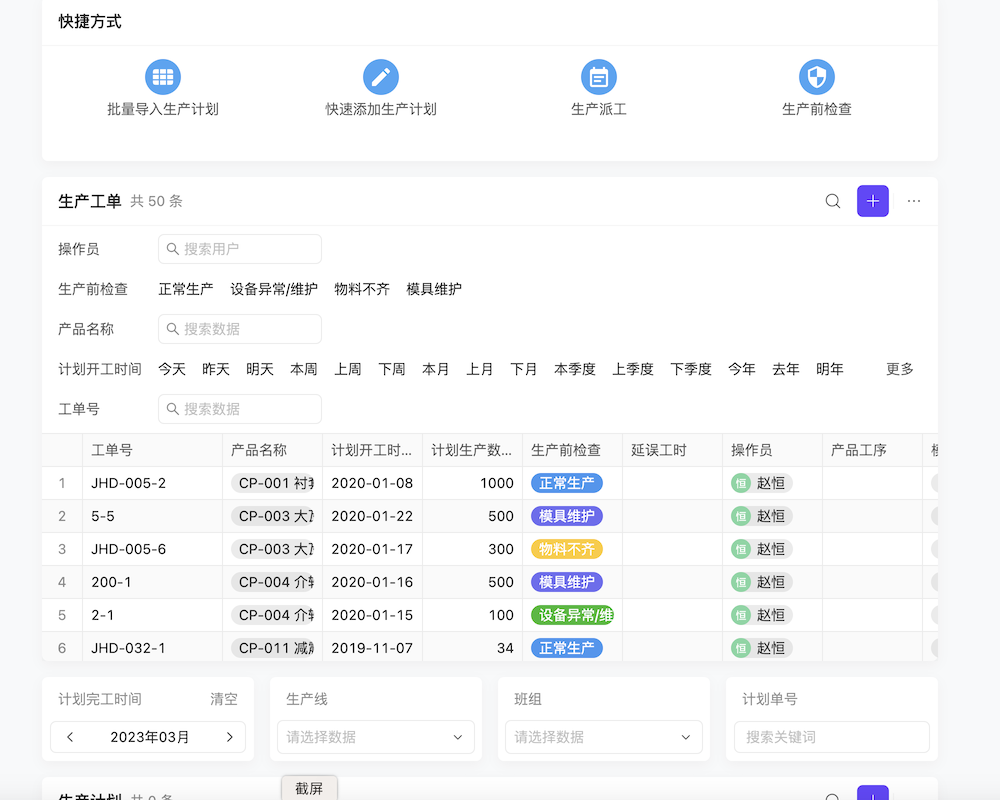
医疗器械产品管理办法对生产企业提出了多项具体要求。首先,生产企业需要建立并维护质量管理体系,确保产品生产过程的可追溯性和质量控制。其次,必须严格遵守生产环境、设备、原材料等方面的规定,确保产品符合安全有效标准。此外,还需加强员工培训,提高员工的质量意识和操作技能。同时,生产企业还需配合监管部门的监督检查,及时整改发现的问题。遵守这些规定,不仅有助于提升企业形象,更能保障公众用械安全。想要了解更多生产企业合规要求吗?点击我们的平台免费注册试用吧!
医疗器械产品不良事件如何监测与报告?
万一我家的医疗器械出了啥问题,比如用了之后有不良反应,这该怎么监测和报告呢?
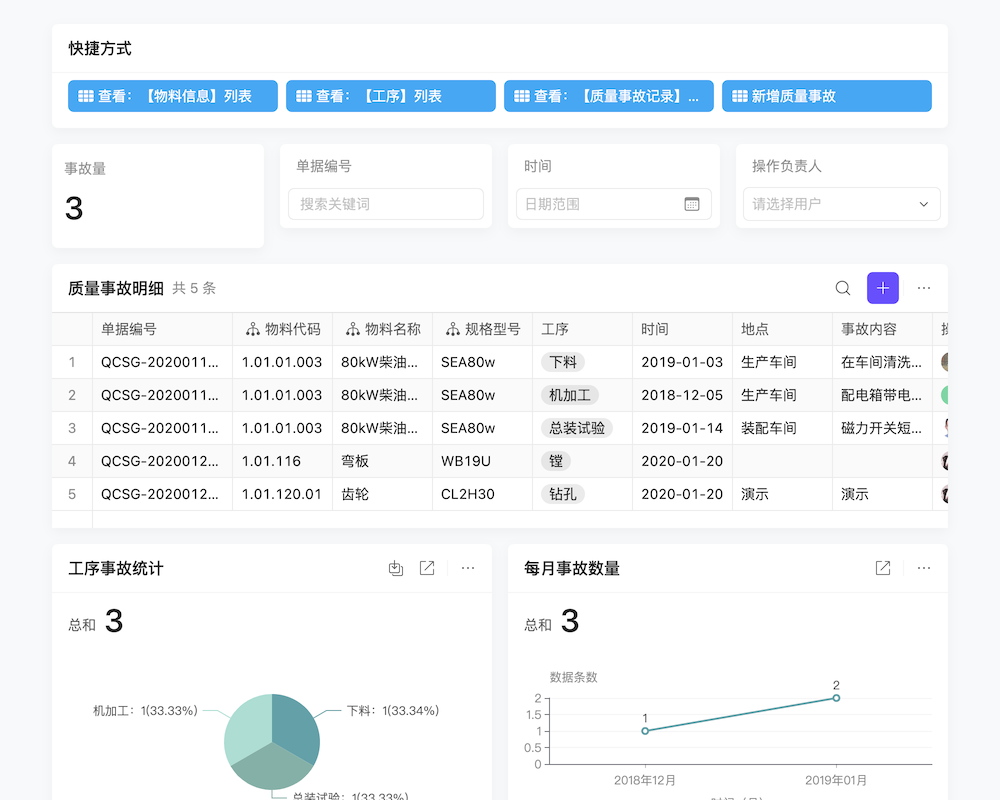
医疗器械产品不良事件的监测与报告是保障公众用械安全的重要环节。生产企业应建立不良事件监测制度,明确监测职责与流程。一旦发现不良事件,应立即进行详细记录,并初步分析事件原因。随后,需按照规定的时限和途径向监管部门报告,同时积极配合监管部门的调查与处理。此外,生产企业还应加强产品召回管理,对于存在安全隐患的产品,应及时启动召回程序,确保问题产品不再流入市场。想要更好地掌握不良事件监测与报告的技巧吗?不妨预约我们的专业演示,获取更多实战经验分享哦!
免责申明:本文内容通过 AI 工具匹配关键字智能整合而成,仅供参考,伙伴云不对内容的真实、准确、完整作任何形式的承诺。如有任何问题或意见,您可以通过联系 12345@huoban.com 进行反馈,伙伴云收到您的反馈后将及时处理并反馈。